Medical Device testing and compliance solutions to meet state-of-the-art standards on safety and electromagnetic compatibility (EMC)
Intertek provides expert safety testing and electromagnetic compatibility (EMC) solutions, helping medical device manufacturers meet critical global standards with greater efficiency, speed, and confidence. From design to market entry, our services ensure compliance with IEC 60601-1, IEC 60601-1-2, and other key regulations for safety, EMC, and medical device performance.
With expertise in medical imaging technologies, implantable devices, home healthcare equipment IEC 61010 for laboratory equipment and much more, we address the unique challenges of modern medical innovations. Our advanced facilities and experienced engineers provide reliable testing that ensures your devices meet the highest standards of safety, performance, and reliability.
Intertek helps you navigate complex compliance requirements and accelerate time to market, delivering confidence that your devices perform safely in real-world environments. We are your trusted partner to bring innovative, compliant, and high quality medical solutions to the global healthcare market.
Intertek's Medical Device Testing Solutions

Overview of IEC 60601-1 Standards and References
End-to-end solutions from product development and risk management file reviews to comprehensive testing to the IEC 60601-1 series.

IEC 60601-1-2: Medical Device EMC Testing
IEC 60601-1-2 Electromagnetic Compatibility (EMC) Testing for Medical Devices including IEC 60601-1-2 4th Edition Amendment 2.

In Vitro Diagnostic (IVD) Medical Device Testing & Certification
Intertek provides comprehensive testing, certification, and regulatory guidance for In vitro diagnostic (IVD) medical devices throughout the entire product lifecycle.

Home Healthcare Equipment Testing and Certification
Comprehensive Testing and Certification solutions for Home Healthcare Equipment to all applicable standards including IEC 60601-1-11.
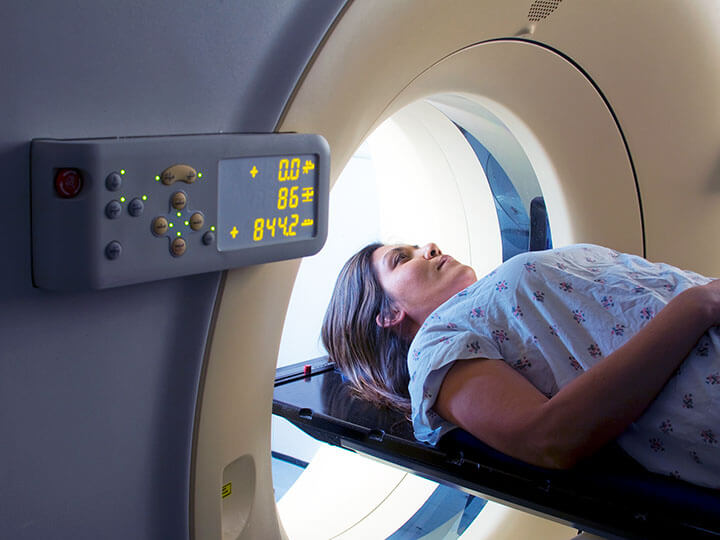
Medical Imaging Equipment Testing
Expert Medical Imaging Equipment Testing and Certification solutions ensuring your medical imaging equipment meets all applicable safety requirements.
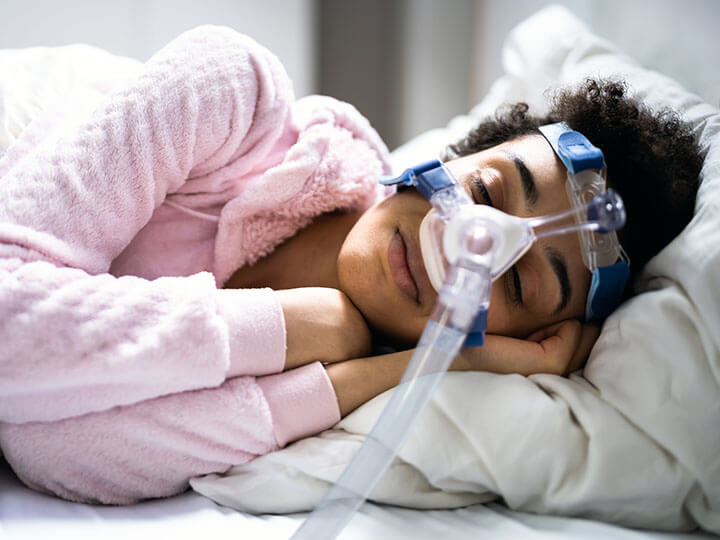
Biocompatibility Testing and Evaluation
Unrivaled capabilities in Biocompatibility Testing and Evaluation for medical devices, delivering comprehensive solutions aligned with ISO standards to ensure safety and compliance.
ISO 18562 and VOC Testing for Medical Devices
The industry leader in ISO 18562 and VOC Testing and Evaluation solutions of breathing gas pathways in medical devices with comprehensive expertise and solutions.

Medical Laboratory Equipment: IEC 61010 and Electrical Safety Requirements
Critical information on IEC 61010, the standard for testing Medical Laboratory Equipment, to ensure your products remain in compliance with the latest revision of IEC 61010.

Implantable Medical Devices Testing Solutions
Guidance on innovative Implantable Medical Devices such as Cochlear Implants, monitoring devices, pacemakers, etc. to comply with safety, EMC and product-specific standards.
Ventilator Production & Testing
Immediate Ventilator Testing assistance for manufacturers along with regulatory requirement guidance for safety, performance, labeling and more.
SPE-3000: Medical Field Evaluations
For medical products entering the Canadian market, SPE-3000 serves as the model code for the field evaluation of medical electrical equipment (MEE) and medical electrical systems (MES), specifically pertaining to safety from electric shock, fire and mechanical hazards.
SATELLITE™ Client Test Program For Medical Devices
Product compliance testing in your own labs and on your own schedule while obtaining our market-leading Certifications.
Preliminary Design Reviews For Medical Devices
A Preliminary Design Review (PDR) enables you to design with confidence and streamline compliance for your medical device.
Medical Device Testing Frequently Asked Questions (FAQs)
Medical device testing is the process of evaluating medical products to ensure they are safe, effective, reliable, and compliant with regulatory standards before they reach patients or healthcare environments. It involves a wide range of assessments; mechanical, electrical, biological, chemical, software, and usability to confirm that a device performs as intended under normal and foreseeable conditions. Testing may include biocompatibility, sterilization validation, electrical safety, electromagnetic compatibility (EMC), software verification, usability studies, and performance or durability evaluations.
Regulatory bodies such as the FDA (in the United States), the EU MDR (in Europe), and other global authorities require comprehensive testing data as part of the approval or certification process. Through these evaluations, manufacturers can identify potential risks, verify design requirements, and ensure that devices meet the strict standards needed to protect patient health and safety.
Medical device testing is important because it protects patient safety, ensures device effectiveness, and supports compliance with strict regulatory requirements. Medical devices interact directly with the human body or clinical environments, so any failure, whether electrical, mechanical, biological, or software-related, can lead to serious harm. Testing helps identify risks early, verifies that the device performs reliably over its intended lifespan, and ensures it can withstand real-world conditions such as repeated use, sterilization, or electromagnetic exposure.
Regulators like the FDA, EU MDR authorities, and other global agencies require extensive testing data to approve devices for market release. Thorough testing also builds manufacturer confidence, reduces liability, and supports product innovation by confirming that design choices meet safety and quality standards. Ultimately, medical device testing is essential for safeguarding patients, supporting clinicians, and maintaining trust in healthcare technology.
Ensuring the safety, performance, and regulatory compliance of medical devices requires adherence to a broad range of international standards and legal frameworks. These standards guide manufacturers through testing, documentation, and product validation to ensure devices are safe for patients and effective in clinical use.
Electrical Safety & Electromagnetic Compatibility
- IEC 60601 Series – Electrical Medical Equipment
- IEC 60601-1 – General electrical safety
- IEC 60601-1-2 – Electromagnetic compatibility (EMC)
- IEC 60601-1-x – Collateral standards (e.g., alarms, usability, radiation protection)
- IEC 60601-2-x – Particular standards for specific device types (e.g., ventilators, infusion pumps)
These standards ensure devices operate safely in clinical environments and do not interfere with other medical equipment.
Software & Cybersecurity Standards
- IEC 62304 – Software Lifecycle Processes
Specifies development, maintenance, and risk controls for medical device software. - IEC 82304-1 – Health Software Safety
Applies to standalone medical software and health applications. - Cybersecurity Guidance
- FDA cybersecurity expectations for secure design, patching, and vulnerability management
- ISO 81001-5-1 for health software security engineering
Mechanical & Performance Testing Standards
A wide range of device-specific standards apply to specialized products, including:
- ASTM F88 – Seal strength testing
- ASTM F1925 – Tensile testing for materials
- ISO 80369 – Small-bore connector safety
- ISO 25539 – Vascular implant requirements
These tests verify durability, structural integrity, and real-world performance.
Quality & Risk Management Standards
- ISO 13485 – Quality Management Systems
Defines the quality system requirements for medical device design, manufacturing, and distribution. - ISO 14971 – Risk Management
Provides a structured approach for identifying, evaluating, and controlling risks throughout a device’s lifecycle.
Biocompatibility & Biological Safety Standards
- ISO 10993 Series – Biological Evaluation
Covers testing for cytotoxicity, sensitization, irritation, systemic toxicity, genotoxicity, implantation, and more. - Sterilization & Microbiology
- ISO 11137 – Radiation sterilization validation
- ISO 11135 – Ethylene oxide sterilization validation
- ISO 11737 – Bioburden testing, sterility assurance, and microbiological evaluation
Packaging, Transportation & Sterile Barrier Systems
- ISO 11607 Series
Requirements for packaging that maintains sterility through shipping and shelf-life. - ASTM & ISTA Standards
- ASTM D4169 – Distribution and shipping testing
- ASTM F1980 – Accelerated aging for shelf-life validation
- ISTA 2A/3A – Transportation simulation programs
Usability & Human Factors
- IEC 62366-1 – Usability Engineering
Ensures device interfaces are intuitive and minimize use-related risks. - FDA Human Factors Guidance
Provides requirements for validation studies and user-interface evaluations.
Regulatory Frameworks by Region
- United States – FDA Regulations
- 21 CFR Part 820 – Quality System Regulation
- 21 CFR Part 807/814 – Device approvals (510(k), PMA)
- Compliance supported by FDA-recognized consensus standards
- European Union – MDR / IVDR
- EU 2017/745 (MDR) – Medical Device Regulation
- EU 2017/746 (IVDR) – In Vitro Diagnostics Regulation
- Requires CE marking and conformity assessment through Notified Bodies.
- Global Frameworks
- IMDRF – International Medical Device Regulators Forum
- Country-specific authorities such as Health Canada, Japan PMDA, China NMPA, and Australia TGA
Determining which tests a medical device needs begins with understanding its regulatory classification and intended use. Every device is assigned a risk class—such as FDA Class I, II, or III in the United States or Classes I through III under the EU MDR—which directly influences the level of testing required. Higher-risk devices typically require more extensive evaluations, including performance testing, biocompatibility assessments, electrical safety, EMC testing, and often clinical evidence. Manufacturers must also consider how the device interacts with the body, including whether it is used externally, inserted, implanted, or in contact with blood or tissue. Factors such as duration of contact, sterility requirements, and whether the device contains electronics or software all play a major role in determining the necessary test plan.
Material selection is another key driver of testing needs. The composition of polymers, metals, coatings, adhesives, and electronic components can trigger requirements for biocompatibility, chemical characterization, mechanical strength testing, and extractables and leachables analysis. In addition, ISO 14971 risk management principles help identify possible hazards—such as electrical shock, breakage, software malfunction, or user error—and map them to the appropriate standards, including IEC 60601 for electrical safety, IEC 62304 for software, IEC 62366 for usability, and ASTM or ISO device-specific performance standards. Packaging, sterilization, distribution, and shelf-life considerations further influence the testing framework, especially for sterile or single-use products.
Medical device testing supports regulatory approval by providing the objective evidence regulators need to confirm that a device is safe, effective, and compliant with all applicable standards. Agencies such as the FDA, EU MDR Notified Bodies, Health Canada, and others cannot authorize a device for market unless the manufacturer demonstrates—through validated test data—that it performs as intended and poses minimal risk to patients or users. Testing results form the backbone of the technical documentation required for submissions like 510(k)s, PMAs, CE Marking technical files, and global market approvals.
Testing also verifies that a device meets recognized standards, including ISO 10993 for biocompatibility, IEC 60601 for electrical safety and EMC, ISO 11607 for sterile packaging, IEC 62366 for usability, and IEC 62304 for medical software. Compliance with these standards streamlines the review process by giving regulators confidence that the device aligns with established safety and performance benchmarks. Additionally, testing provides essential evidence for risk management under ISO 14971, demonstrating that known hazards have been identified, evaluated, mitigated, and validated through design verification and validation activities.
Medical device testing can be performed both in-house and through an accredited third party, depending on the type of evaluation and your regulatory needs. Many manufacturers conduct early design verification and pre-compliance testing internally to refine prototypes and identify issues before formal certification. However, most global regulators and market access schemes require an independent, accredited laboratory, such as Intertek, to perform or validate compliance testing before a product can be certified or placed on the market. Third-party evaluation helps ensure impartiality, consistency, and traceable results that meet the expectations of regulators, Notified Bodies, and certification agencies.
For manufacturers with strong internal laboratory capabilities, Intertek offers its SATELLITE data acceptance program that allows you to perform certain tests in your own lab while still benefiting from Intertek’s accreditation, oversight, and certification expertise. Under this program, qualified client labs can execute approved test plans in-house, with Intertek reviewing the data and issuing the final certification. This approach gives you the flexibility to accelerate timelines, maintain greater control over your test schedule, and keep sensitive prototypes onsite, while still meeting the requirements for independent verification.
Related Links

Ensuring Safety in Electrically Powered Patient Lifts | White Paper
Electrically powered patient lifts must be safe, reliable, and compliant. This whitepaper explores the dual compliance requirements of IEC 60601-1 (electrical safety) and ISO 10535 (mechanical safety and performance), helping manufacturers avoid common pitfalls and streamline approvals.

Creation of IEC 60601-1 4th Edition
Stay ahead in medical device compliance with our exclusive webinar on the IEC 60601-1 4th Edition updates. Learn about critical changes, streamlined standards, and how to future-proof your designs for global markets.
Knowledge Center
Download the latest information from our medical device compliance experts.
Six Medical Device Trends Shaping 2026
Six Strategies for Medical Device Compliance in 2026
Medical EMC In-Situ Testing: On-demand Webinar | White Paper
IEC 81001-5-1 and Cybersecurity for Medical Devices Webinar
Six Compliance-Related Questions About Connected Home Healthcare Devices
Accelerated Stress Testing for Medical Devices
Machine Learning and Artificial Intelligence (AI) in Medical Devices: Webinar | Fact Sheet
IEC 60601-1-2 Ed. 4.1 Overview of Requirements
Medical OEM Wireless Coexistence Testing
Biocompatibility Risk Assessment and Evaluation Plans
For more expert papers, recordings, and presentations, visit our Medical Resources hub.


Follow Us For More!
We’re always adding new content and looking for ways to help you simplify the regulatory and compliance process for medical devices.
*The Intertek legal entities that provide medical device management system certification services (including ISO 13485 and MDSAP) and Notified Body services (MDR 2017/745 and MDD 93/42/EEC) do not provide any consulting services. Clients who have used other Intertek legal entities’ consulting services are not eligible to receive management system certification services or Notified Body services from Intertek.
